TCGA预后基因联合免疫浸润
TCGA预后基因联合免疫浸润通过ESTIMATE算法计算免疫,间质和ESTIMATE得分,并以此筛选DEGs,进行生存分析、KEGG、GO、GSEA分析,构建PPI网络并筛选hub
通过ESTIMATE算法计算免疫,间质和ESTIMATE得分,并以此筛选DEGs,进行生存分析、KEGG、GO、GSEA分析,构建PPI网络并筛选hub基因,最后通过构建风险评分(RS)模型,研究hub基因的预后价值并用验证集进行验证。
题目:从TCGA数据库中筛选对急性髓系白血病有预后价值的基因
一.研究背景
急性髓系白血病(AML)是一种造血克隆性恶性肿瘤。AML患者的治疗和预后取决于准确的细胞遗传学和基因检测。AML疾病状况的改善主要得益于支持疗法和造血细胞移植的进步。然而,由于高复发率,AML的5年生存率仍然很低,因此迫切需要新颖有效的治疗方法。从免疫治疗的角度出发,希望找到与AML的肿瘤微环境相关的具有预后价值的基因。
二.分析流程
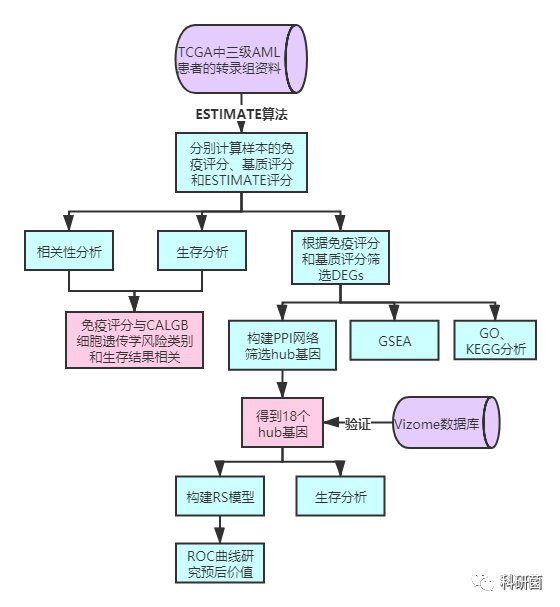
三.结果解读
1.数据收集与处理
收集了TCGA中三级AML患者的转录组资料,即入选的样本在样本ID中以"-03"结尾,属于"原发性血源癌-外周血"。一共入选了173例样本。使用GDC工具提取样本的临床特征和生存数据,使用limma包进行归一化处理。
2. 免疫评分与癌症和急性白血病B组(CALGB)细胞遗传学风险类别和生存结果相关
使用ESTIMATE算法分别计算样本的免疫评分、基质评分和ESTIMATE评分。绘制箱线图,观察上述评分与CALGB细胞遗传学风险类别的关系。
免疫评分越高与CALGB细胞遗传学风险类别的风险等级越高相关(P=0.0396,图1A)。
基质评分和ESTIMATE评分未观察到与CALGB细胞遗传学风险类别之间的显著性(分别为P = 0.8585和P = 0.3320,图1B,C)。
根据免疫评分、基质评分和ESTIMATE评分的中位数分别将AML样本分为高分组和低分组,绘制生存曲线,评估这三个评分与总体生存率之间的关系。
免疫评分和ESTIMATE评分越高,总体生存情况越差(分别为P=0.0224,P=0.0195,图1D,F)。
基质评分的不同水平与总体生存率之间无显著关系(P=0.3676,图1E)。
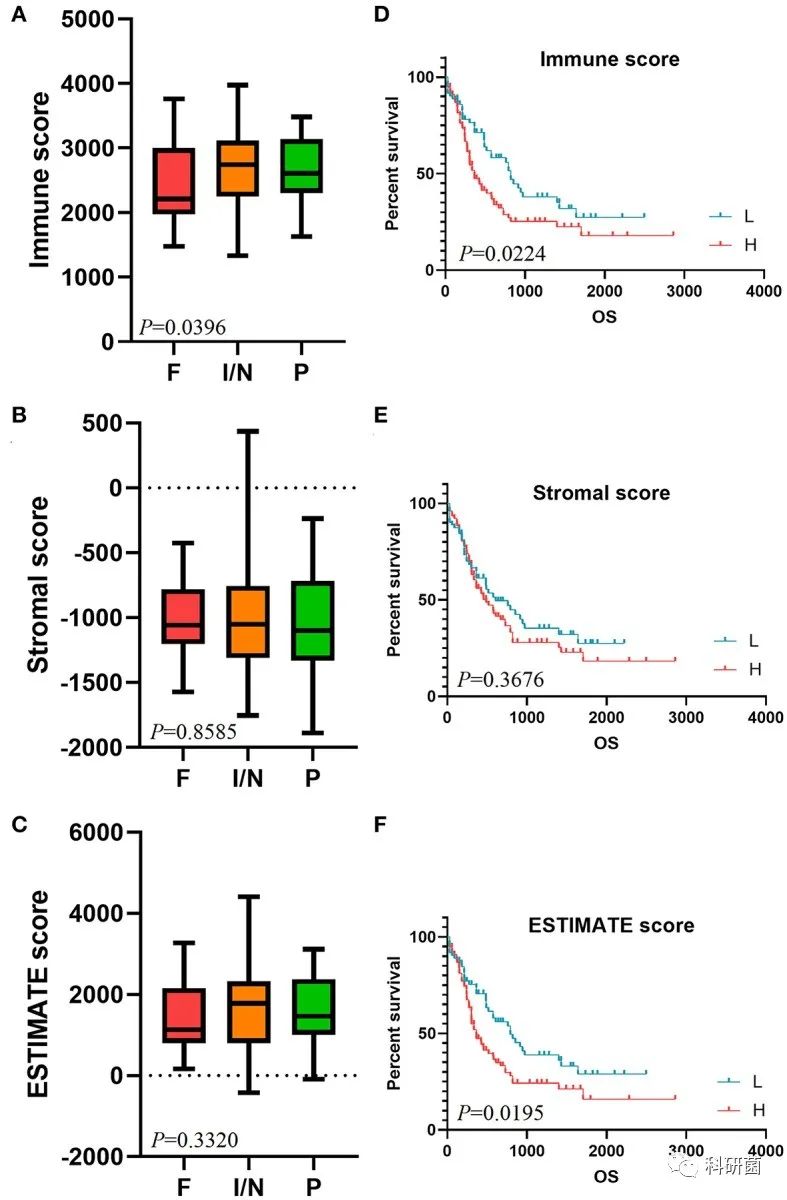
图1 免疫评分、基质评分、ESTIMATE评分在CALGB细胞遗传学风险类别中的分布情况箱线图以及生存曲线
3.AML中基于免疫评分和基质评分的表达谱比较
将免疫评分和基质评分按中位数分为高组和低组,分别绘制基因表达热图(图2A,图3A)。并根据免疫评分和基质评分的高低组分别筛选DEGs(PFDR < 0.05 , |log2FC| > 1),绘制火山图(图2B,图3B)。通过Venn图对两组获得的DEGs取交集。
在免疫评分组中筛选了331个上调的DEGs和889个下调的DEGs。
在基质评分组中筛选了195个上调的DEG和870个下调的DEGs。
在Venn图中,筛选出147个有交集的上调基因(图2C)和680个有交集的下调基因(图2D)。
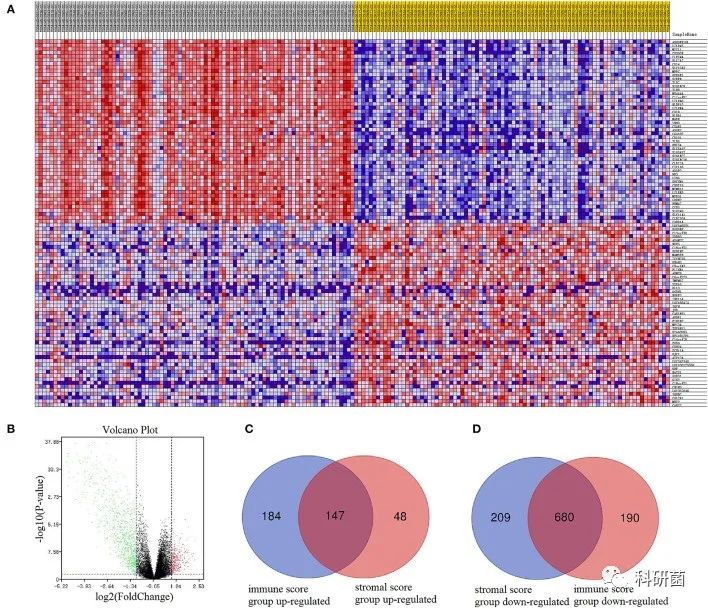
图2 基于免疫评分分组的表达谱热图、火山图以及Venn图
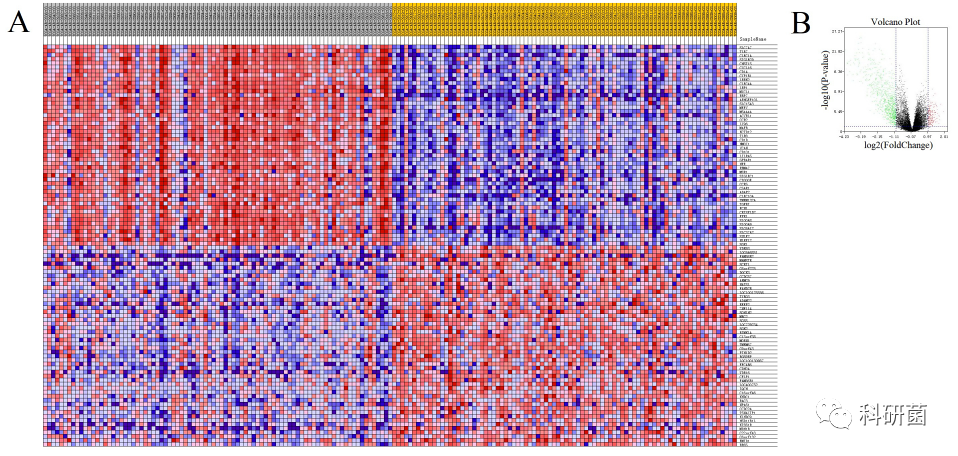
图3 基于基质评分分组的表达谱热图、火山图
4.功能富集分析
对上一步得到的有交集的DEGs进行GO和KEGG富集分析。结果显示这些DEGs和免疫反应存在显著关联。
GO分析主要富集于炎症反应、免疫反应、细胞膜、受体活性。图4A,B,C分别展示了生物过程、细胞成分和分子功能的前10个条目。
在KEGG通路注释(图4D)和富集分析(图4E)中,主要是与免疫、癌症和结核病相关的通路。通路富集分析的前20条结果如图4F所示。
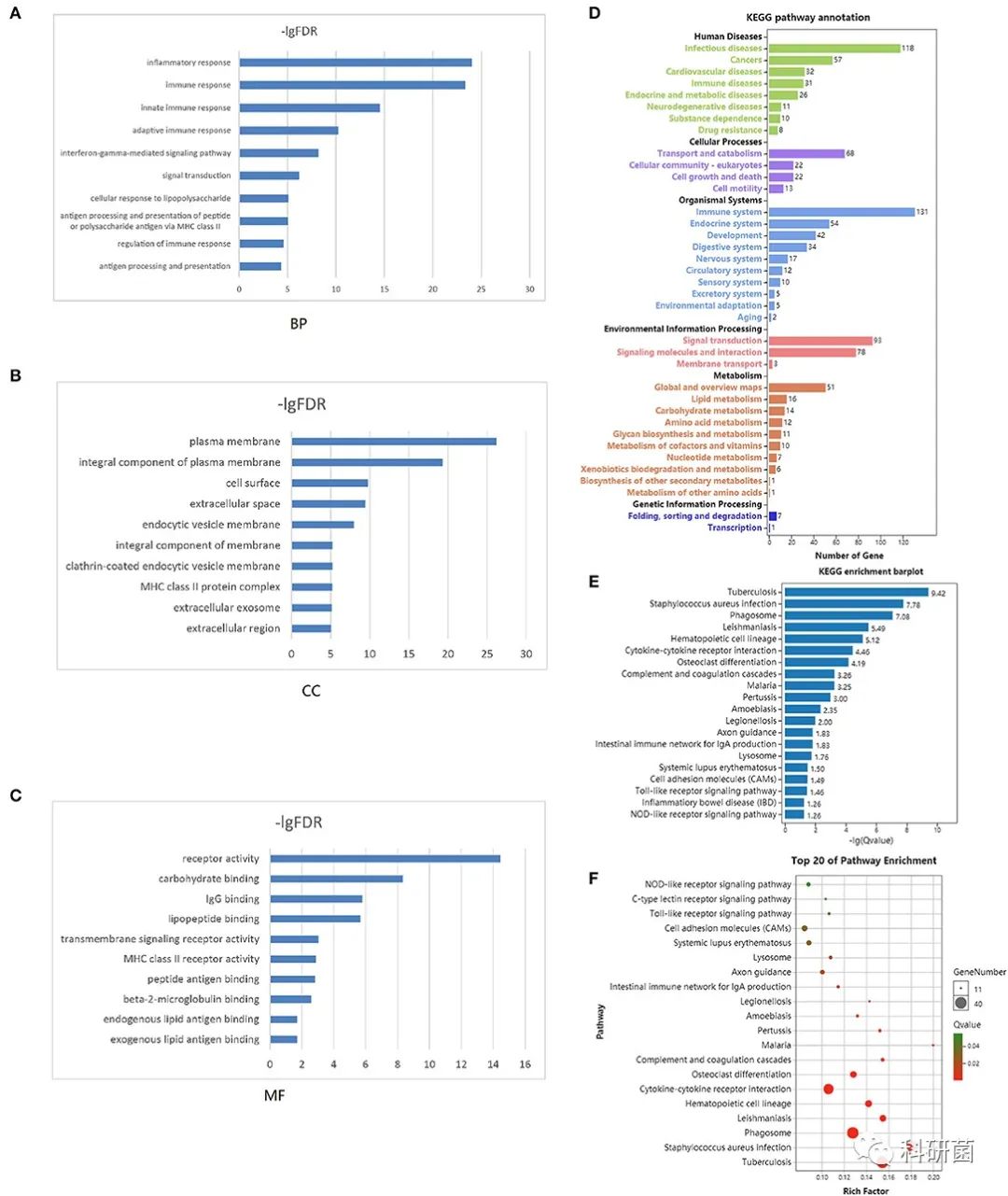
图4 GO和KEGG分析结果
5.PPI网络的构建以及hub基因筛选
使用STRING数据库构建了包含786个节点和1774条边的PPI网络。使用Cytoscape的cytoHubba插件来识别hub基因,从12种算法中选择了前10个节点,并排除了degree<10的基因。最后,确定了18个TME相关的hub基因:ITGAL、ITGAM、HLA-DRB1、HLA-DRB5、FPR1、CX3CR1、TNFRSF1B、CXCL16、CTSB、CTSS、HLA-DRA、P2RY13、ITGB2、CEACAM3、SLC11A1、C5AR1、ADORA3和GNGT2。
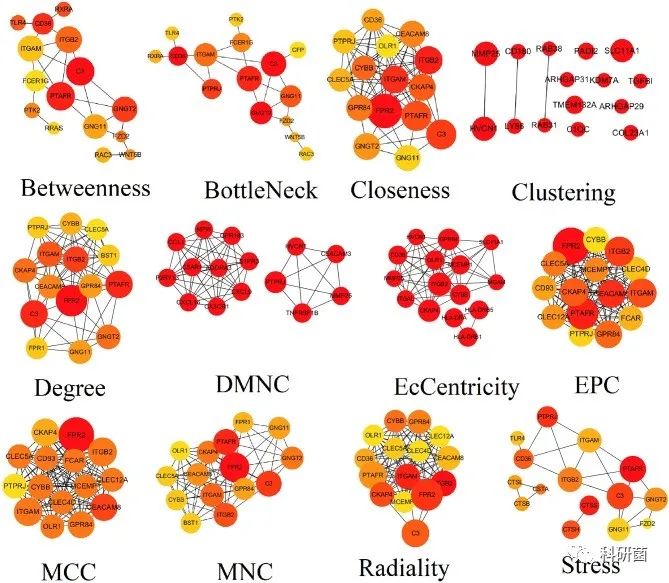
图5 cytoHubba插件基于12种算法进行hub基因识别
6.GSEA分析
随后进行GSEA分析,并设置FDR<0.25、|enriched score|> 0.35、基因集内基因数≥35。
GSEA的结果表明:抗原加工和呈递,B细胞受体信号传导通路,趋化因子信号传导通路,FcγR介导的吞噬作用,移植物与宿主疾病,造血细胞谱系,产生IgA的肠道免疫网络,NK细胞介导的细胞毒性,NOD样受体信号传导通路,T细胞受体信号传导通路和Toll样受体信号传导通路是肿瘤免疫相关交叉基因富集的主要通路(图6)。
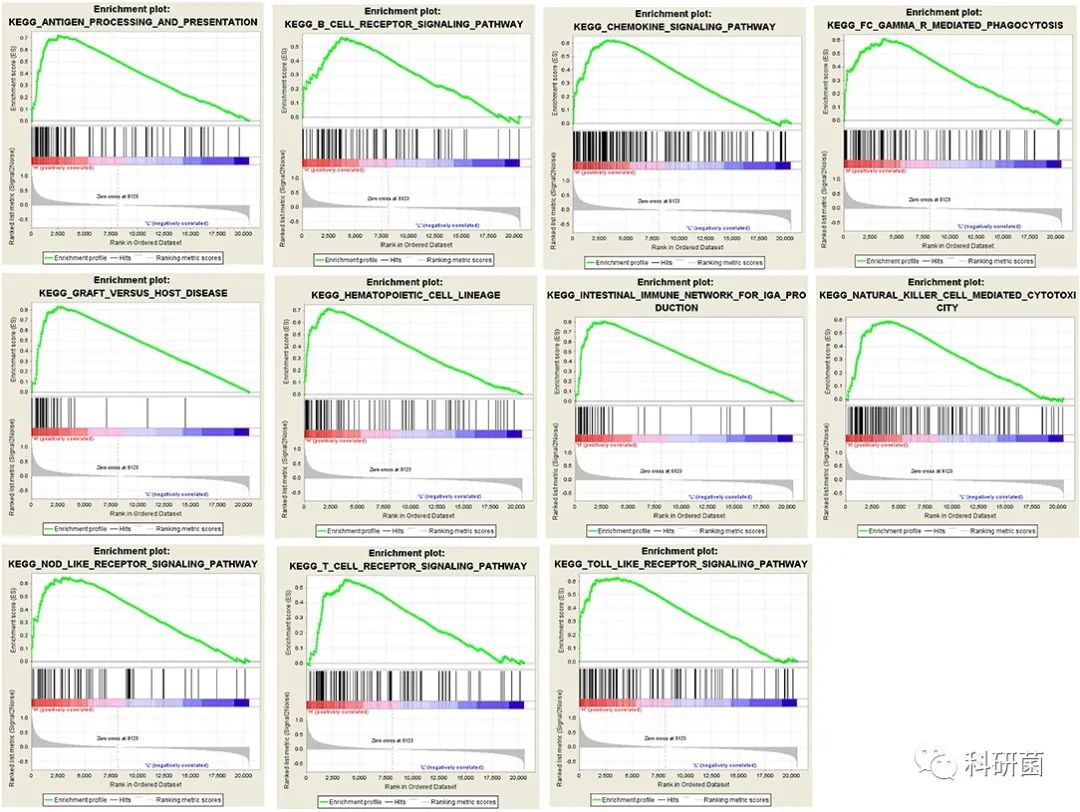
图6 GSEA分析的结果
7.风险评分和生存分析
基于多变量Cox回归分析,计算出每个AML患者的风险评分(RS)公式。
RS = ITGAL * 0.177 + ITGAM * 0.315 + HLA-DRB1 * 0.371 + HLA-DRB5 * (?0.009) + FPR1 * 0.034 + CX3CR1 * (?0.074) + TNFRSF1B * 0.172 + CXCL16 * (?0.104) + CTSB * (?0.38) + CTSS * (?0.201) + HLA-DRA * (?0.353) + P2RY13 * 0.003 + ITGB2 * 0.038 + CEACAM3 * (?0.051) + SLC11A1 * (?0.034) + C5AR1 * (?0.049) + ADORA3 * 0.213 + GNGT2 * 0.208
随后根据中位数将163例AML患者分为低RS组和高RS组,进行生存分析,研究不同RS水平与总体生存率之间的关系。结果表明,高RS与不良的总生存期有关(图7A)。然后绘制ROC曲线,并计算曲线下面积AUC为0.725,显示出RS对总体生存率的预测准确性较高(图7B)。
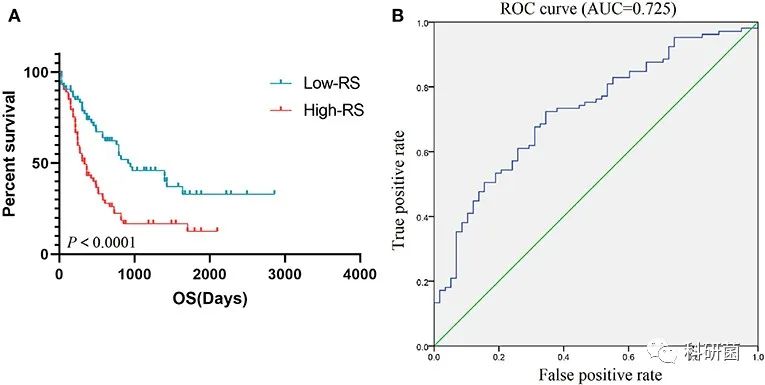
图7 RS的预后价值
此外,还绘制了18个hub基因的生存曲线,以探讨预后价值(图8)。结果显示,hub基因的高表达水平与不良的总体生存率有关。
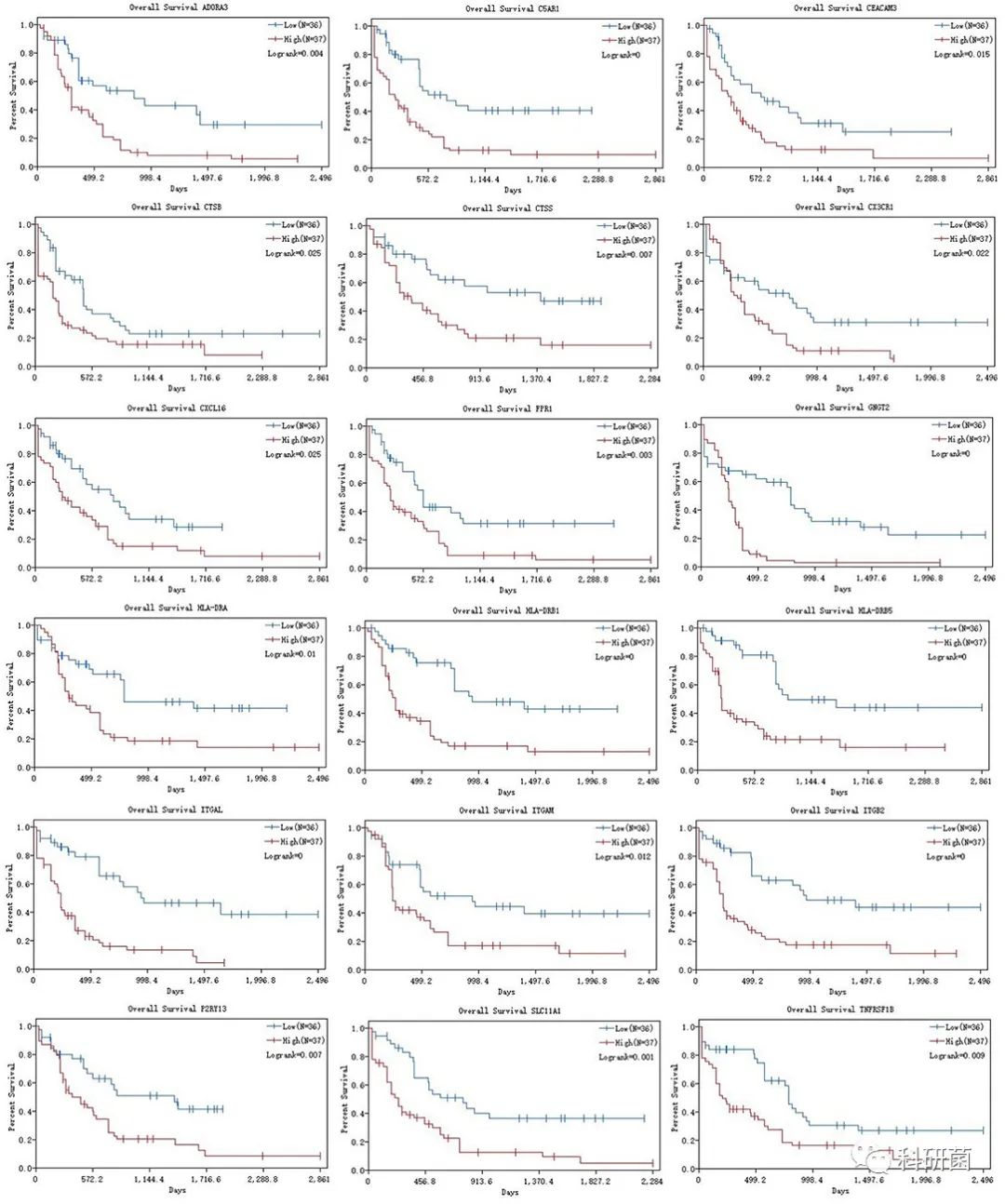
图8 18个hub基因的K-M曲线
8.Vizome数据库分析
Vizome是最大的AML数据库,它包含了从562名患者收集的672份肿瘤样本的全外显子组测序数据。验证了Vizome数据库中hub基因的表达水平。热图显示,18个hub基因在数据库中的样本中表现出高表达(图9A)。图9B中还显示了其中4个hub基因的相互作用关系。
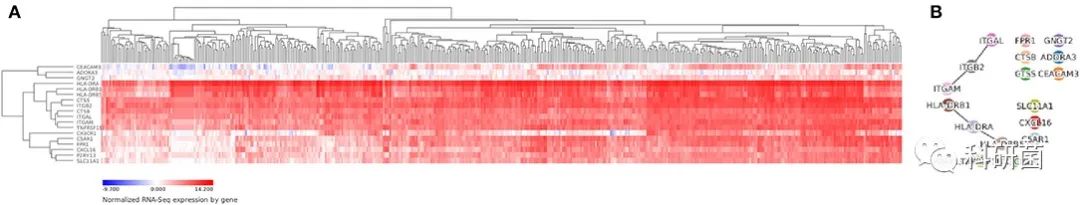
图9 验证Vizome数据库中hub基因的表达水平
小结
利用TCGA数据库,基于ESTIMATE算法得出的免疫评分和基质评分得到了可预测AML患者预后不良的TME相关基因,并对其进行对进行功能富集分析。构建PPI网络,筛选得到TME相关的hub基因,并构建RS模型,为预测AML患者的生存状况提供了新的依据。
-
哪种蓝牙耳机最好?评分最高的五款口碑耳机2020-01-16
-
中国移动智能硬件质量报告:2500-3500元荣耀20 Pro评分第一2019-06-28